
WSolids1: Solid-State NMR Simulation
 | Current Release: 1.21.7 (09.07.2021) Contents: [General] - [Calculation Models] - [Changes] - [Availability] |
General:
- A 32 bit program that runs under MS Windows XP and above to simulate solid-state NMR spectra (it does not iterate). WSolids1 also runs under Linux using Wine or under Mac OS, also using a variant of Wine
- Save everything as a self-contained WSolids document (in XML format). In connection with your web browser, this XML file can be used to generate a report.
For security reasons, the XML file and the required XSL and CSS helper files need to reside in the same directory. New distributions of WSolids1 will bundle these helper files in a subdirectory "report" of the WSolids1 program directory. If you cannot write to this subdirectory, maybe you can copy them to a location where you have write access, or you can download the helper files here: report.zip (please refer to the help file). - For detailed information, have a look at the PDF documentation (2.1 MB).
- Calculations typically take a few seconds to complete.
- An experimental spectrum can be read in one of the following formats: Bruker (TopSpin, XWinNMR, WinNMR, WinNMR ASCII), Chemagnetics Spinsight, JCAMP-DX, Simpson, and a native ASCII format.
- Calculated spectra can be displayed in absorption, first derivative, or second derivative mode.
- Output of calculated spectra is in TopSpin, WinNMR, JCAMP-DX or Solids format. WinNMR, TopSpin or a similar program (e.g., SpecPlot) is used for plotting.
  | WSolids1 can also serve to interface with Budzelaar's gNMR, or Perras Automatic Multiple Experiment Simulation and Fitting AMES-fit: to get TopSpin spectra into gNMR and back, as outlined on a separate page: gNMR |
Supported Calculation Models:
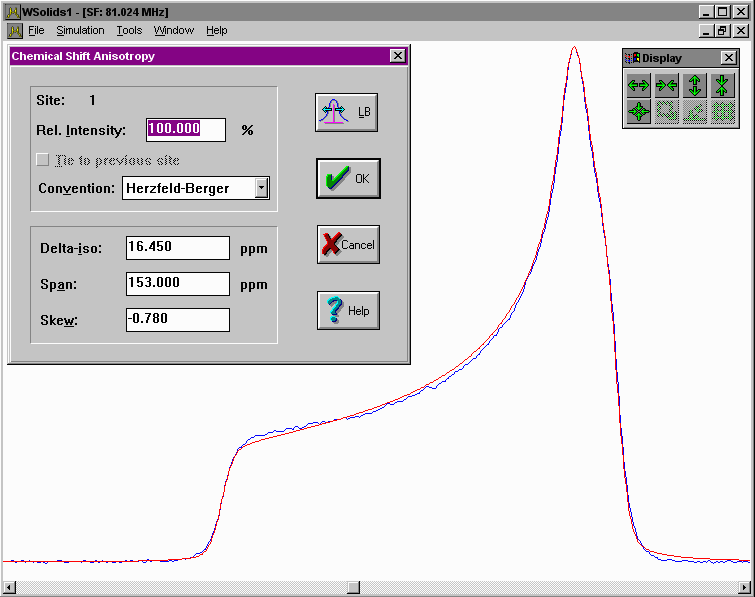 | Static: Chemical shift anisotropy Spectrum of a static powder sample showing chemical shift anisotropy ("powder pattern"). |
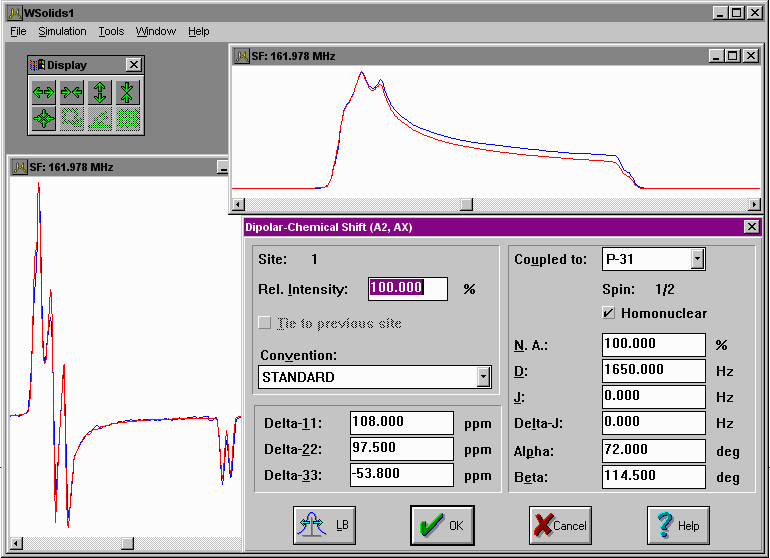 | Static: Dipolar-chemical shift (A2, AX) Chemical shift anisotropy, direct dipole-dipole coupling and indirect spin-spin coupling for a homonuclear pair of equivalent spin-1/2 nuclei (A2 approximation) or a heteronuclear spin pair in a static powder sample (AX approximation). |
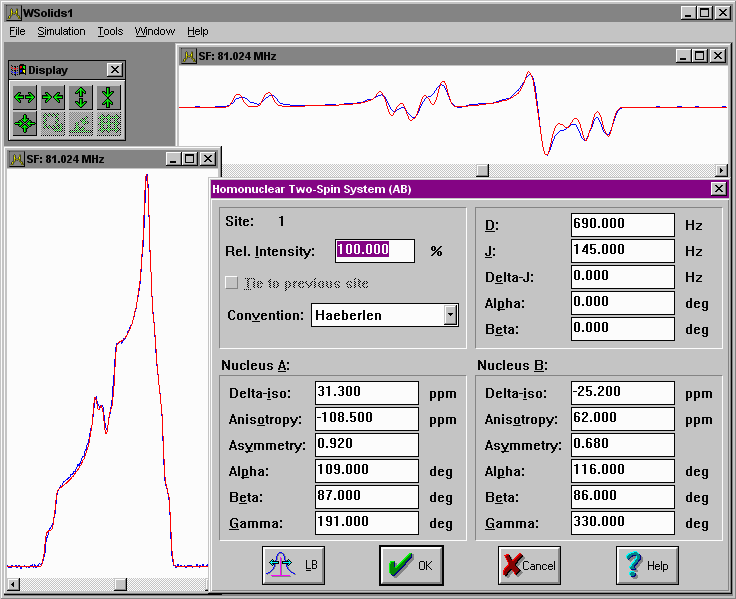 | Static: Dipolar-chemical shift (AB) Chemical shift anisotropy, direct dipole-dipole coupling and indirect spin-spin coupling for a homonuclear pair of spin-1/2 nuclei, including "second-order" effects, in a static powder sample (AB). |
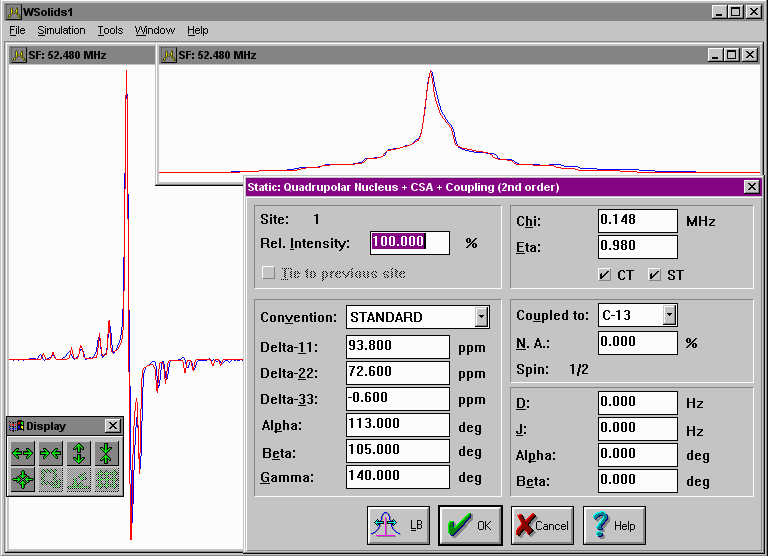 | Static: Quadrupolar nucleus Quadrupolar interaction up to second order for the observed nucleus, including chemical shift anisotropy, for a static powder sample. Optionally, dipolar and indirect coupling to a heteronucleus can be added (note: quadrupolar interaction, if any, is neglected for the coupled heteronucleus). |
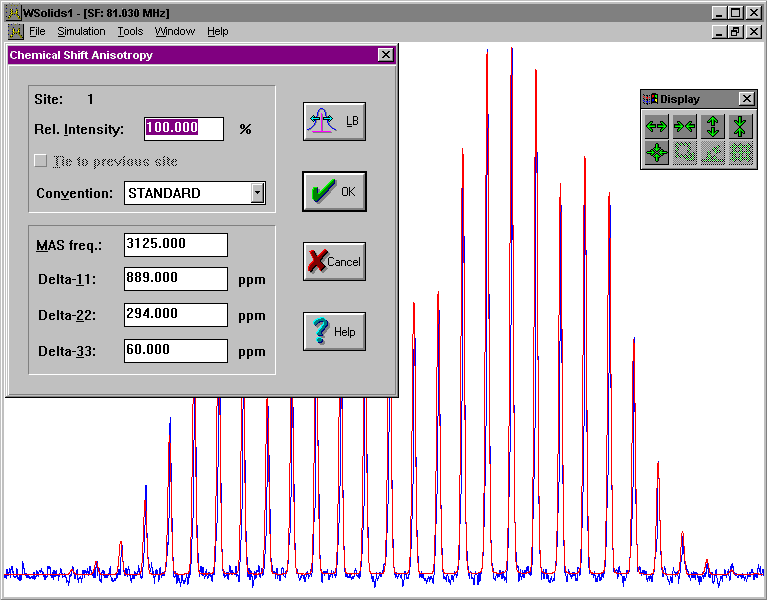 | MAS: Chemical shift anisotropy (HB) Spectrum of a powder sample spinning at the magic angle, showing chemical shift anisotropy; uses Herzfeld-Berger tables, if applicable. |
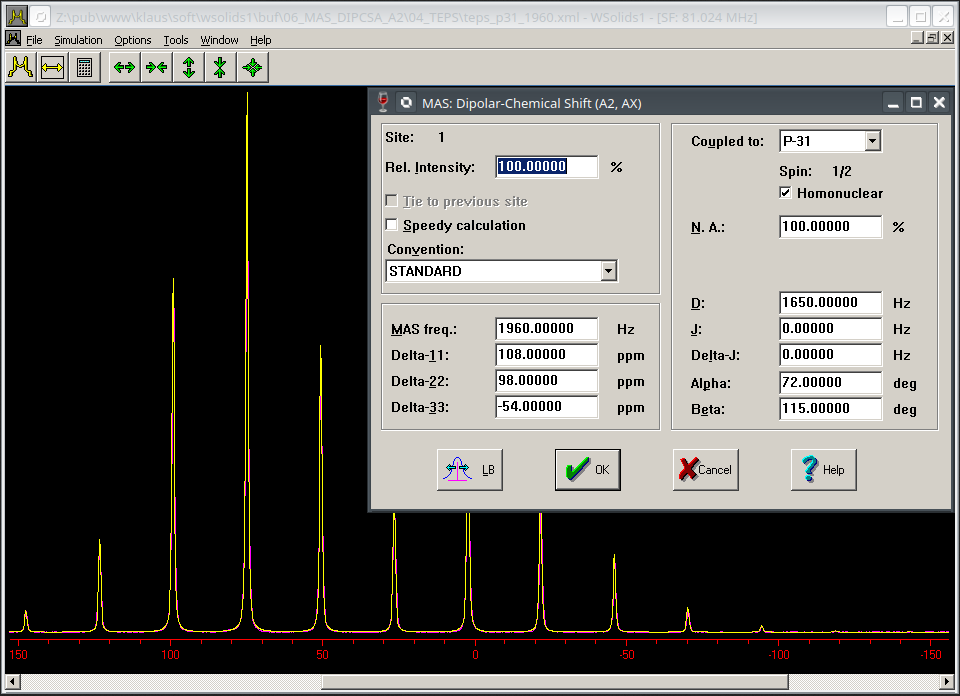 | MAS: Dipolar-chemical shift anisotropy (A2, AX) Chemical shift anisotropy, direct dipole-dipole coupling and indirect spin-spin coupling for a homonuclear pair of equivalent spin-1/2 nuclei (A2 approximation) or a heteronuclear spin pair (AX approximation) in a powder sample under magic angle spinning. |
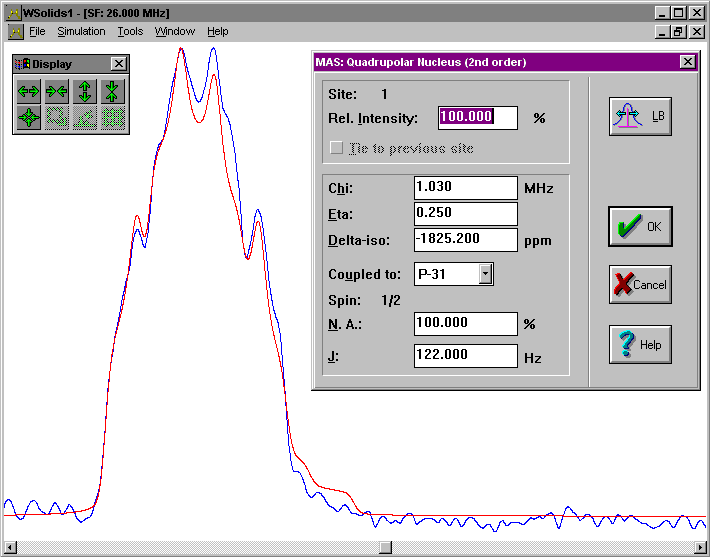 | MAS: Quadrupolar nucleus Spectrum of central transition of a quadrupolar nucleus in a powder sample spinning rapidly at the magic angle. |
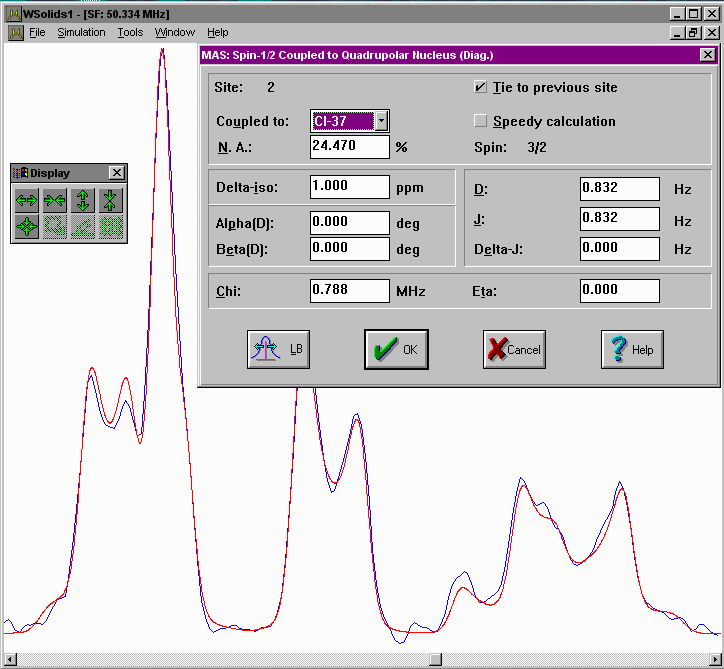 | MAS: Spin-1/2 -- Spin-S (Diag.) Considers spin-spin interactions with a quadrupolar nucleus under magic-angle spinning, using full matrix diagonalization. |
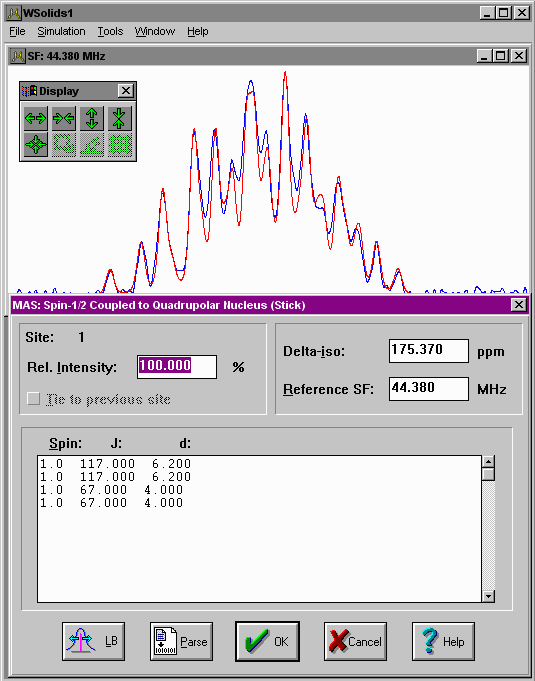 | MAS: Spin-1/2 -- Spin-S (Stick) Considers spin-spin interactions with quadrupolar nuclei under magic-angle spinning, using first-order perturbation theory and "stick" approach. |
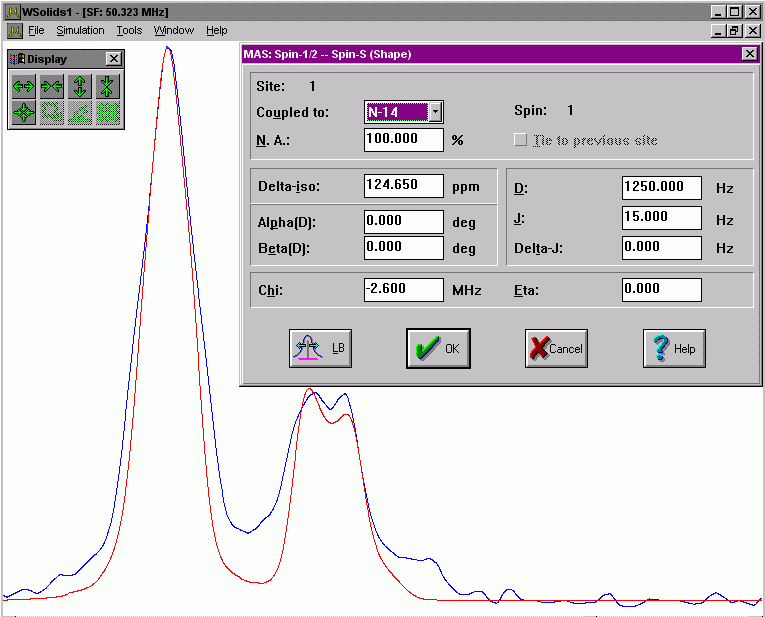 | MAS: Spin-1/2 -- Spin-S (Shape) Considers spin-spin interactions with a quadrupolar nucleus under magic-angle spinning, using first-order perturbation theory to calculate the line shape. |
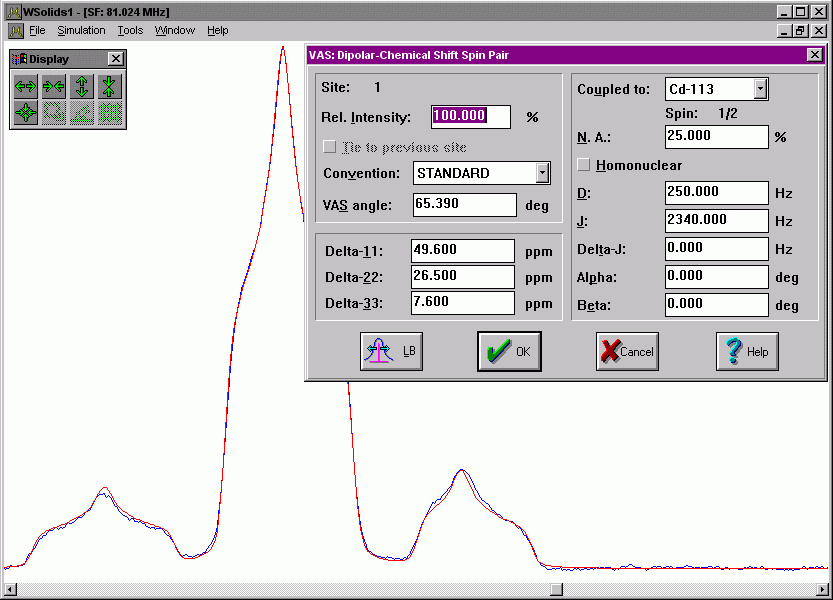 | VAS: Dipolar-chemical shift (A2, AX) Considers chemical shift and spin-spin interactions for a homo- or heteronuclear pair of nuclei, i.e. A2 or AX approximation, under variable-angle spinning (fast spinning limit). |
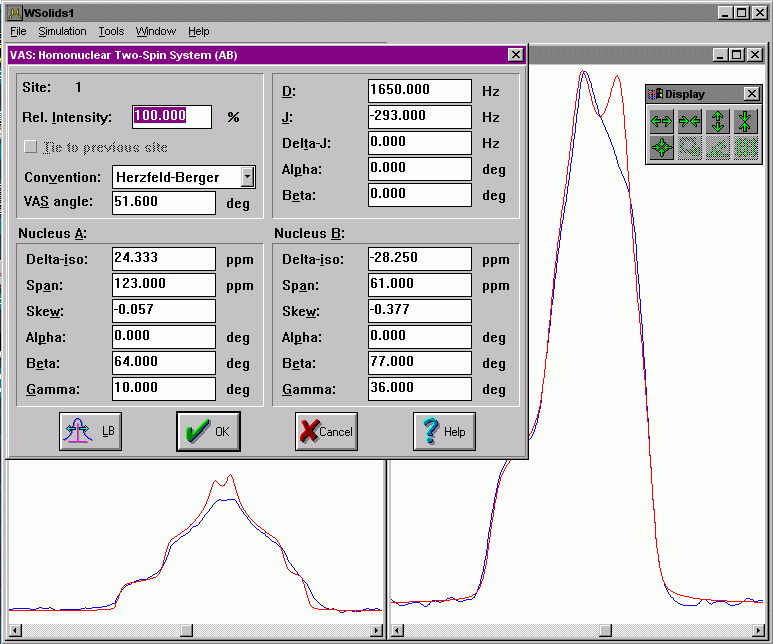 | VAS: Dipolar-chemical shift (AB) Considers chemical shift and spin-spin interactions for a homonuclear pair of nuclei, i.e. AB approximation, under variable-angle spinning (fast spinning limit) |
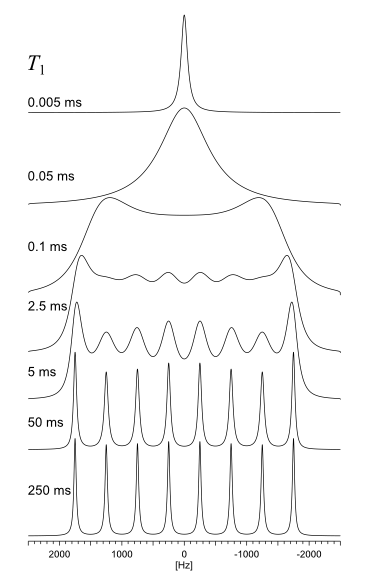 | HR: Spin-1/2 Coupled to Quadrupolar Nucleus (Relaxation) Actually a high resolution, solution spin system, a spin-1/2 nucleus coupled to a quadrupolar nucleus where the spin-lattice relaxation of the quadrupolar nucleus affects the spectrum of the spin-1/2 nucleus. |
Changes in 1.21.7 (09.07.2021):
- new feature: in the MAS: Chemical Shift Anisotropy (HB) and MAS: Dipolar-Chemical Shift (A2, AX) models, new Herzfeld-Berger tables are used to allow for more spinning sidebands and greater spans: spinning sideband orders in the range from -30 to +30, and values of μ in the range 0 ≤ μ ≤ 50 are now possible (compared to ±15 ssbs and μ ≤ 30); for parameters outside the Herzfeld-Berger tables, WSolids1 does a direct calculation
- new feature: the reading of JCAMP-DX has been extended to accept NTUPLES (e.g. produced by TopSpin 3 and 4); also some errors dealing with XY-DATA or with malformed JCAMP-DX files (e.g. exported from Vnmrj) have been fixed (thanks to reports by Ulla Gro Nielsen, Andrew Root, Robbie Iuliucci, Ernest Prack, Danielle Laurencin).
- Neither bug nor feature: determination of the Adobe Acrobat version looks up now a registry key to determine the proper version of the Acrobat DDE server
Availability
The program is available at no cost if you write me an email. The installation file is about 3.3 MB in size.
If you find this program useful and publish results obtained using this program, please include a reference to this program similar to the following (depending on the journal style):
WSolids1 ver. 1.21.7, K. Eichele, Universität Tübingen, 2021.